Introduction¶
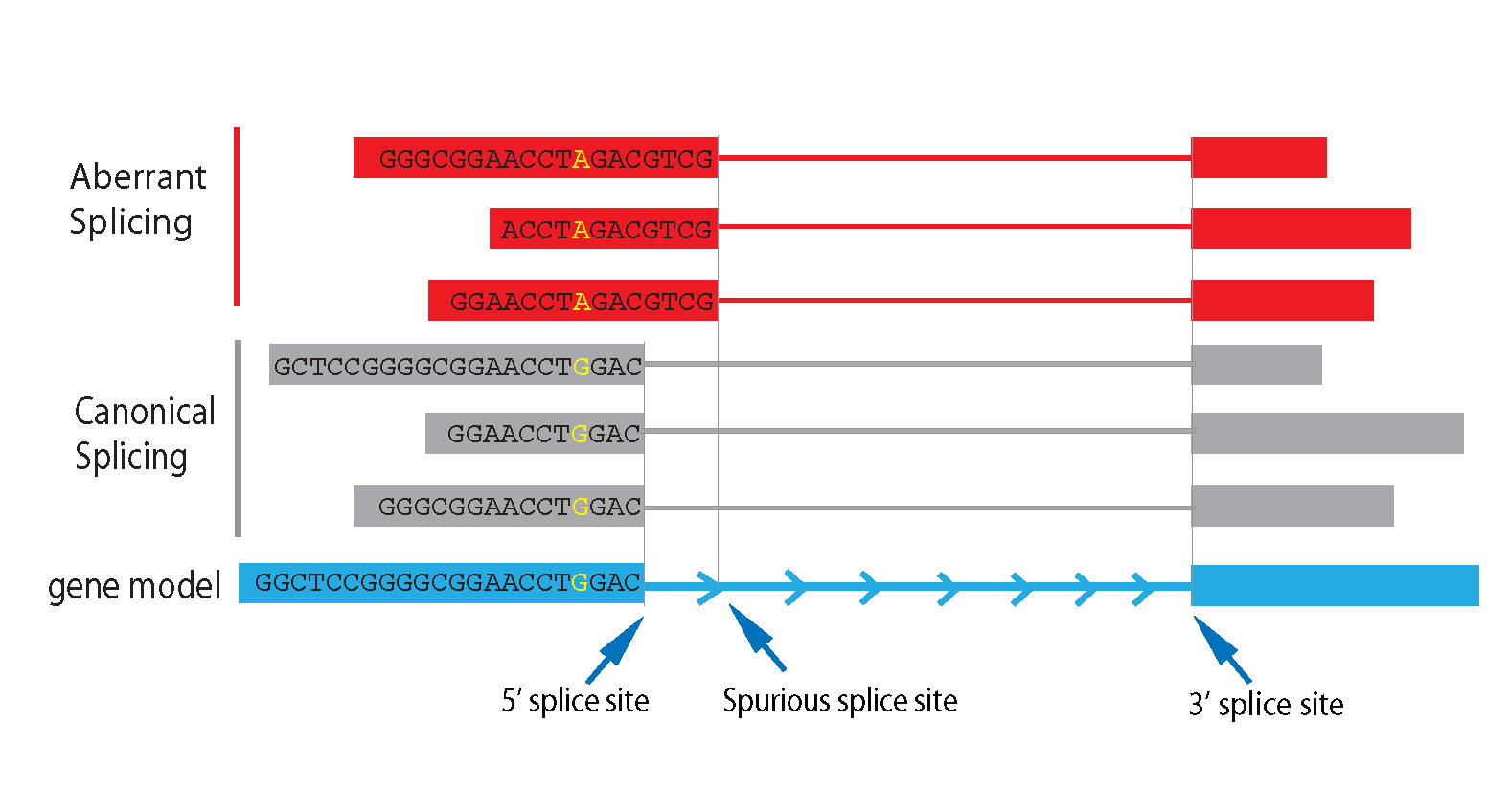
Alternative splicing is a precisely regulated process that involves interactions between cis- and trans-acting regulatory elements. Factors (such as genetic mutation) that disrupt these interactions result in aberrant splicings. Deep transcriptome sequencing (RNA-seq) has the capability to interrogate both “aberrant splicing variants” and “sequence variants (SNP, indel, etc)” simultaneously, thus provides the unique opportunity to identify “sequence variants” that directly associated with aberrant splicing. According annotation status, splicings junctions detected from RNA-seq can be divided into 3 categories:
- Canonical splicing: both 3’ splice site (3’SS) and 5’ splice site (5’SS) are known.
- Semi-canonical splicing: only one of the 3’SS and 5’SS is known, the other splice site is novel.
- Novel splicing: both 3’SS and 5’SS are novel.
We define semi-canonical splicing and novel splicing as aberrant splicing, as both of them use spurious splicing sites. PVAAS (Program to identify Variants Associated with Aberrant Splicing) is such program to identify single nucleotide variants and indels that are associated with aberrant splicing. As shown in example below, G->A mutations are strongly associated with the usage of 5’ spurious splicing site.
Installation¶
Prerequisites¶
- gcc
- python2.7.*
- numpy and scipy
- cython
- Xcode (MacOS only)
Check dependencies¶
# check gcc
$ gcc --version
Configured with: --prefix=/Applications/Xcode.app/Contents/Developer/usr --with-gxx-include-dir=/usr/include/c++/4.2.1
Apple LLVM version 5.0 (clang-500.2.79) (based on LLVM 3.3svn)
Target: x86_64-apple-darwin12.5.0
Thread model: posix
# check python version
$ python --version
Python 2.7.2
# check numpy and scipy
python -c 'import numpy'
python -c 'import scipy'
# check cython
cython --version
Cython version 0.19.2
Install¶
$ tar zxf PVAAS-VERSION.tar.gz
$ cd PVAAS-VERSION
$ python setup.py install
# or you can install PVAAS to a specified location:
$ python setup.py install --root=/some/where
Usage¶
Usage: pvaas.py [options]
- Options:
--version show program’s version number and exit -h, --help show this help message and exit -i FILE_BAM, --input=FILE_BAM BAM file. BAM file should be indexed and sorted using SAMtools. -g GENE_BED, --gene=GENE_BED Referencde gene models in BED format. Must be standard 12 column. File can be plain text or gzipped. -r GENOME_FA, --genome=GENOME_FA Reference genome sequence file in FASTA format. File can be plain text or gzipped. ‘-r’ and ‘-v’ are mututally exclusive. -v VARIANT_VCF, --vcf=VARIANT_VCF VCF file containing user-defined variant. File can be plain text or gzipped. ‘-v’ and ‘-r’ are mututally exclusive. -o OUTPUT_PREFIX, --output=OUTPUT_PREFIX Prefix of output file. default=pvaas_output -q MIN_MAPQ, --mapq=MIN_MAPQ Minimum mapping quality. Mapping quality is Phred-scaled probability of the alignment being wrong. default=30 -m MIN_SEQQ, --seqq=MIN_SEQQ Minimum sequence quality. Sequence quality is Phread-scaled probability of the base calling being wrong. default=30 -p P_VAL, --pvalue=P_VAL Fisher exact test pvalue (adjusted using benjamini- hochberg procedure) cutoff. default=0.05 -d MIN_DIST_SIZE, --dist=MIN_DIST_SIZE Minimum distance between variant and the termini of read. default=5 -c MIN_COVERAGE, --cvg=MIN_COVERAGE Minimum number of reads supporting one allele. default=2 -k, --clean Remove intermediate ‘.sortName.bam’ -f, --vcffilter Turn on VCF filter (only varaint marked with “PASS” will be used.
Results¶
PVAAS generates these files
prefix.spliceFrag.bam¶
This BAM file contains splice alignments extracted from the original BAM file. For single-end RNA-seq, only the alignment records of spliced reads were extracted; For pair-end RNA-seq, if one read were spliced, the alignment records of both reads (read1 and read2) would be extracted from the original BAM file for downstream analysis. Each alignment was annotated using the provided gene model (i.e. BED file specified by ‘-g’), in particular, three different tags (“KK”,”KN”,”NN”) were appended to each splice read (and its mate) as:
- KK (Known-Known): Both 5’ splice site (5’SS) and 3’ splice site (3’SS) are annotated (named canonical splicing). You might see multiple “KK” tags if read was spliced multiple times or both ends were spliced. ( eg: KK:Z:chr1:14829-14969.)
- KN (Known-Novel): One splice site is known, the other splice site is novel (named semi-canonical splicing). (eg. KN:Z:chr1:783186-784863).
- NN (Novel-Novel): Both splice sites are novel (named novel splicing).
Example:
UNC14-SN744:268:C10DDACXX:4:1308:13818:35728/2 163 chr9 14660 50 48M = 14929 457 CTTCCGCTCCTTGAAGCTGGTCTCCACCCACTGCTGGTTCCGTCACCC 8=?D:DDDHAHDBE@HGIBHE@<D9<E3)):C3BD<?0:?######## RG:Z:120904_UNC14-SN744_0268_AC10DDACXX_4_GTGGCC IH:i:1 HI:i:1 NM:i:1 KK:Z:chr9:14940-15080
UNC14-SN744:268:C10DDACXX:4:2201:9246:148959/1 99 chr9 173290 66 48M = 175773 4465 ACCGTTTCTAAGTTCCAGCCACTCTTCATAGAGCTCTCCACCTTGGCT CCCFFFFFHHHHFIJJJJJIJJJJJJJJJIJIJIIJJJIJJJIIIJJJ RG:Z:120904_UNC14-SN744_0268_AC10DDACXX_4_GTGGCC IH:i:1 HI:i:1 NM:i:0 KN:Z:chr9:175784-177718
UNC14-SN744:268:C10DDACXX:4:2103:5415:27906/2 163 chr9 2769934 55 48M = 2770128 7813 CAAATGCTGGGATTACAGATGTGAACCACCATGCCCAGCTAGAGGCTC CCCFFFFFHHGHBHIJJJJJJJJJIJJIJJJJJJJJJJJJGIHIJIIH RG:Z:120904_UNC14-SN744_0268_AC10DDACXX_4_GTGGCC IH:i:1 HI:i:1 NM:i:1 NN:Z:chr9:2770163-2777734
You would see the combination of “KK”, “KN” and “NN” if the read (pair) has multiple annotation status. For example, one read is canonical splicing while its mate is semi-canonical splicing.
prefix.rawPval.xls¶
Spreadsheet (11 columns) containing variants that are potentially associated aberrant splicing. P values were not adjusted and filtered.
- Chromosome name.
- Chromosome position (1-based).
- Reference allele.
- Allele frequency (AF) of all reads. All reads were counted without filting.
- Allele frequency (AF) of reads supporting canonical splicing. Canonical splicing is splicing event that has two known splice sites (i.e. both 5’SS and 3’SS are known). For example, “A174,G67” means 174 reads having allele ‘A’, another 67 reads having allele ‘G’. Only reads passing filter were counted.
- Allele frequency (AF) of reads supporting semi-canonical splicing. Semi-canonical splicing is splicing event that has one known splice site and one novel splice site. Only reads passing filter were counted.
- Allele frequency (AF) of reads supporting novel splicing. Novel splicing is splicing event that has two novel splice sites (i.e. both 5’SS and 3’SS are novel). Only reads passing filter were counted.
- 2x2 table for Fisher Exact test (If the variant was significantly associated with semi-canonical splicing).
- Raw P-value of Fisher exact test(If the variant was significantly associated with semi-canonical splicing).
- 2x2 table for Fisher Exact test (If the variant was significantly associated with novel splicing).
- Raw P-value of Fisher exact test(If the variant was significantly associated with novel splicing).
Example:
chrom position ref_base AF(all) AF(canonical) AF(semi-canonical) AF(novel) 2x2(semi_canonical) raw_pval(semi-canonical) 2x2(novel) raw_pval(novel)
chr9 100823084 T C221,T226 C206,T219 C8,T4 C7,T3 [[219,206],[4,8]] 0.251694519825 [[219,206],[3,7]] 0.212766063103
chr9 37948724 T C33,T38 C30,T35 NA C3,T3 NA NA [[35,30],[3,3]] 1.0
chr9 88631577 G AA3,GA106 AA2,GA103 GA1 AA1,GA2 [[103,2],[1,0]] 1.0 [[103,2],[2,1]] 0.0817805991497
chr9 96320999 C A19,C149 C147 A19 C2 [[147,0],[0,19]] 2.333609442e-25 [[147,0],[2,0]] 1.0
prefix.adjustedPval.xls¶
Format is same as prefix.rawPval.xls. But P values were adjusted using Benjamini-Hochberg procedure. Only variants with adjusted P value < cutoff (specified by -p) were included.
chrom position ref_base AF(all) AF(canonical) AF(semi-canonical) AF(novel) 2x2(semi_canonical) BH_pval(semi-canonical) 2x2(novel) BH_pval(novel)
chr9 96320999 C A19,C149 C147 A19 C2 [[147,0],[0,19]] 1.4001656652e-24 [[147,0],[2,0]] 1.0
Running PVAAS¶
Download these files¶
- hg19.fa.gz (Human reference genome in FASTA format)
- hg19.All.bed12.gz (Human reference gene models in BED format)
- chr9.sorted.bam (Read alignment (BAM) file)
- chr9.sorted.bam.bai (Index file)
run PVAAS with de novo SNP calling¶
python2.7 pvaas.py -i chr9.sorted.bam -r hg19.fa.gz -g hg19.All.bed12.gz -o output
@ 2014-07-15 11:51:20: Running PVAAS v0.1.4
@ 2014-07-15 11:51:20: Loading reference genome hg19.fa
@ 2014-07-15 11:53:28: Reading reference bed file: hg19.All.bed12
@ 2014-07-15 11:53:52: Sorting chr9.sorted.bam by name (read id)
@ 2014-07-15 11:57:27: Save sorted chr9.sorted.bam to output.sortName.bam
@ 2014-07-15 11:57:27: Extracting and annotating splice read and its mate
@ 2014-07-15 11:59:32: Sorting output.spliceFrag.bam by coordinates
@ 2014-07-15 11:59:51: Indexing output.spliceFrag.bam
@ 2014-07-15 11:59:53: Searching for mutation associated with aberrant splicing ...
@ 2014-07-15 13:32:25: Correcting P values using Benjamini & Hochberg's procedure
NOTE:
- Reference genome sequences (i.e. hg19.fa.gz) must be provided to invoke SNP calling.
- PVAAS does not perform indel calling. If users want to associate indels to aberrant splicing, they need to perform indel calling by themselves, then run PVAAS with a VCF file (see below).
run PVAAS with a user defined VCF file¶
python2.7 pvaas.py -i chr9.sorted.bam -v test.vcf -g hg19.All.bed12.gz -o output
@ 2014-11-12 12:22:43: Running PVAAS v0.1.5
@ 2014-11-12 12:22:43: Reading reference bed model: hg19.refseq.bed12
@ 2014-11-12 12:22:45: Sorting chr9.sorted.bam by name (read id)
@ 2014-11-12 12:27:19: Save sorted chr9.sorted.bam to output.sortName.bam
@ 2014-11-12 12:27:19: Extracting and annotating splice read and its mate
@ 2014-11-12 12:29:10: Sorting output.spliceFrag.bam by coordinates
@ 2014-11-12 12:29:41: Indexing output.spliceFrag.bam
@ 2014-11-12 12:30:10: Correcting P values using Benjamini & Hochberg's procedure
Example¶
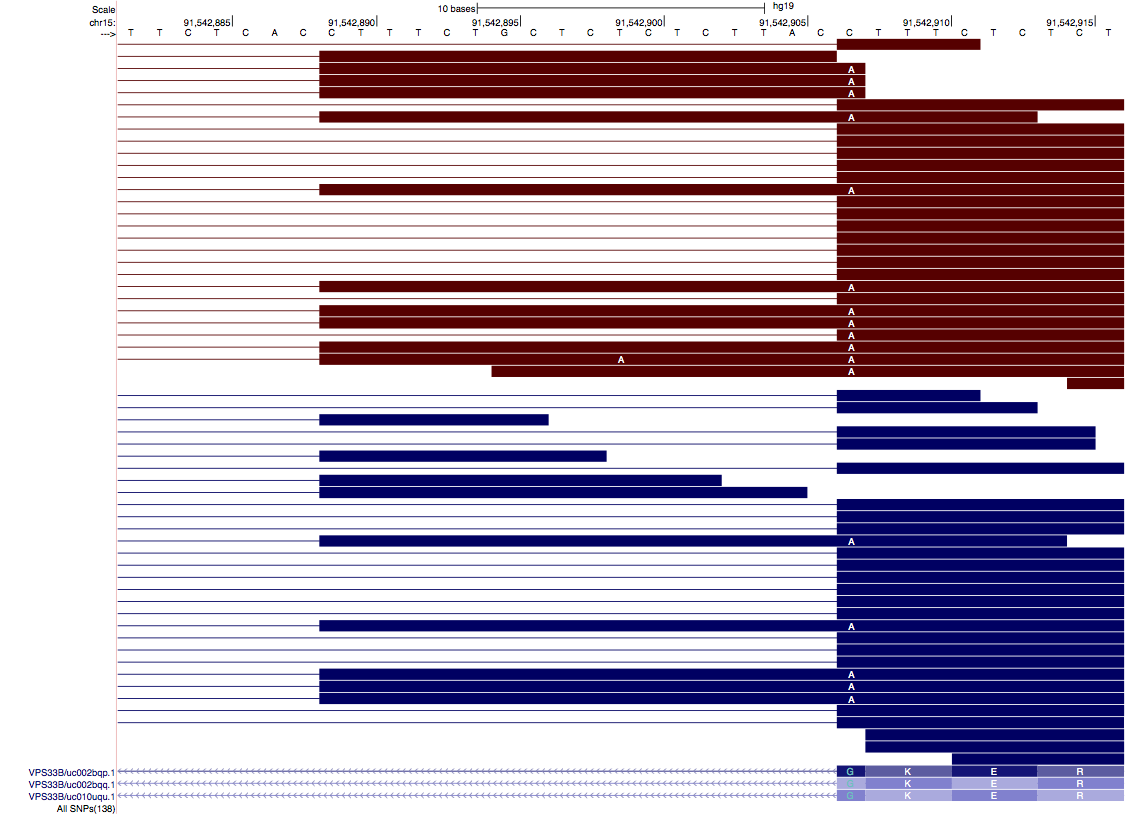
In below screenshot taken from UCSC genome browser, ‘A’ is significantly associated with aberrant splicing (i.e. extended 5’SS). “Blue” bars indicate reads mapped to ‘+’ strand, and “red” bars indicate reads mapped ‘-‘ strand.
Contact¶
Liguo Wang: wangliguo78_AT_gmail.com